Available online at: https://doi.org/10.18778/1898-6773.86.4.05
 https://orcid.org/0000-0002-2481-6050
https://orcid.org/0000-0002-2481-6050
Institute of Legal Medicine, University “Magna Graecia” of Catanzaro, Catanzaro, Italy
https://orcid.org/0000-0002-1750-3742
Institute of Legal Medicine, University “Magna Graecia” of Catanzaro, Catanzaro, Italy
Department of Forensic Medicine, Asl Città di Torino, Turin, Italy
https://orcid.org/0000-0002-0491-7698
Department of Experimental and Clinical Medicine, University “Magna Graecia” of Catanzaro, Catanzaro, Italy
Department of Experimental and Clinical Medicine, University “Magna Graecia” of Catanzaro, Catanzaro, Italy
https://orcid.org/0000-0002-5922-1524
Department of Health Sciences, University “Magna Graecia” of Catanzaro, Catanzaro, Italy
Institute of Legal Medicine, University “Magna Graecia” of Catanzaro, Catanzaro, Italy
https://orcid.org/0000-0001-6637-9402
Archaeology, College of Humanities, Arts and Social Sciences, Flinders University, Adelaide, SA, Australia
FAPAB Research Center, Avola (SR), Sicily, Italy
https://orcid.org/0000-0001-8902-3142
University of Lodz, Faculty of Biology and Environmental Protection, Department of Anthropology, Poland
Institute of Legal Medicine, University “Magna Graecia” of Catanzaro, Catanzaro, Italy
ABSTRACT: Marfan Syndrome (MFS) is an autosomal dominant disease caused in most cases by mutations in the FNB1 gene, which encodes for fibrillin 1. MFS does not alway shows typical phenotypic signs. Indeed, the occurrence of sudden death of unknown cause is increasingly seen in young adults without ante mortem pre-existing pathology to explain the event. In many cases the diagnosis of Marfan Syndrome (MFS) is carried out post mortem, especially in cases where the disease’s external phenotype is absent. Here is reported a case of a young woman who died during a twin pregnancy investigated with medico-legal and forensic anthropological procedures. The autopsy showed the absence of a typical marfanoid habitus and the presence of a dissecting aneurysm of the aorta with histopathological degeneration of the aortic elastic fibers. The genetic investigation revealed two previously undetected de novo mutations of the FBN1 gene: c.T6181C: p.C2061R and c.G1415A: p.C472Y. This new mutations, together with a comprehensive analysis, demonstrates the existence of a causal relationship between these mutations and the dissecting aneurysm of the aorta. This also stresses the importance of a combined multidisciplinary approach to this condition which includes morphological and genetic studies.
KEY WORDS: Marfan Syndrome, genes, mutation, dissecting aneurysm, legal medicine.
Marfan Syndrome (MFS) is an autosomal dominant disorder caused by genetic mutations. In 66-91% of cases, these mutations involve FBN1, a gene that provides instructions for making fibrillin-1 (15q21.1); less frequently, mutations involve TGFβRl or TGFβR2 (also present in Loeys-Dietz syndrome) (Dean 2007). Fibrillin, an extracellular matrix glycoprotein which is a structural component of calcium-binding microfibrils, was discovered in the 1980s thanks to monoclonal antibody technology, whereas FNB1 was directly linked to MFS in 1991 (Sakai et al. 1986). Fibrillins’ domain organisation has been shown to have been conserved throughout evolution, with a structure constituted by 43 calcium-binding epidermal growth factor-like (cbEGF) domains interspersed with 7 transforming growth factor β-binding protein-like (TB) domains (Piha-Gossack et al. 2012; Jensen et al. 2021). Mutated FBN1 alters the structure of fibrillin-1 or decreases its amount, thus compromising the tensile properties of the microfibrils (Frydman 2008). Since, as it will be explained subsequently, no palaeogenetic tests have been carried out so far on possible ancient MFS cases, no information is available regarding the exact first emergence of FBN1 mutations in hominin evolution. Nevertheless, a study by Asgari and colleagues (2020) clearly demonstrated how a positively selected FBN1 missense variant in a Peruvian sample contributes to reducing that population’s stature, which could possibly speak for a major and long-standing role for altered forms of fibrillins in human evolution. Indeed, as Asgari and colleagues underlined (2020) “it is also possible that other FBN1-related traits like changes in cardiovascular system performance have offered an evolutionary advantage” in their studied population.
From a clinico-morphological perspective the disorder can involve several organs and systems: the skeleton (pectus carinatum, pectus excavatum, joint hypermobility, high palate with dental crowding, protrusio acetabuli, scoliosis, reduced elbow extension, pes planus), the eye (ectopia lentis, flat cornea, increased axial length of globe, hypoplastic iris or ciliary muscle), the heart (dilatation of the aortic root, mitral valve prolapse, dissection of the ascending aorta), the lungs (spontaneous pneumothorax, apical blebs), the skin (striae atrophicae) and the dura mater (lumbosacral dural ectasia). These alterations may coexist or not, with a very heterogeneous clinical presentation, which makes the correct diagnosis difficult (Dean 2007). Recent studies by the Milan anatomy research group (Dolci et al. 2018) have once more highlighted the relevance of a morphological assessment of MFS from the facial abnormalities by presenting a quantitative method of assessing the phenotype, whereas other clinical studies focus on the cardiovascular aspects on the disease (Bianucci et al. 2023).
In 2010, the diagnostic criteria for MFS were revised. The two cardinal features of MFS (aortic root aneurysm/dissection and ectopia lentis) assumed a greater role in diagnosing the syndrome and molecular genetic screening for FBN1 and other relevant genes (e.g. TGFβR1 and 2) became more important. Additionally, from the histological point of view, MFS is characterised by loss of elastic fibers, pseudocystic medionecrosis, with deposition of mucoid substances, as evidenced in our case and in other studies (Klintschar et al. 2009). However, these findings are not specific to MFS. Indeed, they are present in other connective disorders (e.g. Ehler-Danlos) and, in the longer term, arterial hypertension, which speaks for the great heterogeneity of this condition, a characteristic which also clearly emerged from external morphological studies (Dolci et al. 2018). Early mortality from MFS results from aortic dilatation (Krause 2000).
These diagnostic and interpretative difficulties must be considered relevant not only to diagnosticians in their attempt at a more refined diagnosis which could help avoid sudden death in MFS patients (i.e. the role of an early diagnosis), but also to (forensic) anthropologists, morphologists and geneticists studying the disease both in modern and ancient populations in an attempt to determine its antiquity and evolutionary course. Hence a multidisciplinary investigation approach ought to be considered optimal for this rare disease which, due to the relative lack of raw date on the quality of life (QOL) of patients, has been the subject of a recent comprehensive study in Poland (Trawicka et al. 2022). This showed how MFS patients have a reduced QOL with subsequent need for medical and psychosocial assistance.
Moreover, Wozniak-Mielczarek et al. (2022) in a morphological study of a Polish population indicated how the diagnosis of MFS still remains “sophisticated” and the differentiation between MFS patients and those who simply present with a marfanoid habitus (i.e. they externally appear to have MFS but do not) cannot be established merely on the basis of an analysis of external body features. It thus follows that the definitive diagnostic tool is genetic analysis, which confirms of the disorder in order to make an early diagnosis and recommend genetic counselling to the patient and their relatives. Genetic counselling allows physicians to give information on the nature, inheritance pattern and implications of the genetic disorder so as to help them make informed medical and personal decisions (Dietz 2001).
For this reason, identifying new mutations linked with MFS and better describing its genotypic background represents a new and fruitful research avenue. In this article we present a recent medical case study and offer both anthropometric, histologic and genetic data, which we believe could be to benefit both of established pathologists and MFS diagnosticians as well as biological anthropologists attempting to apply present-day methodologies to the reconstruction of this antiquity and evolution of MFS.
A 35-year-old woman with a diagnosis of MFS and a positive family history was hospitalised at 23 weeks pregnant with twins because of vomiting and epigastric and spinal pain. She died during an investigation by an angio-CT scan, 4 days after being admitted to hospital. Physicians suspected an aortic dissection to be her cause of death. The patient had undergone cataract surgery years before her death and physicians over time had suspected MFS to be presenting in her. A medicolegal autopsy and a forensic anthropological assessment were carried out. Research of genetic mutations associated with cardiovascular defects was performed. The panel analyses the coding regions of 404 genes related to cardiovascular alterations, as well as 94 noncoding loci implicated in the pathogenesis of cardiovascular diseases and 2 expansion regions of DMPK and CACNA1A genes. The analysis of the sequences focused on coding regions of 16 genes associated with Marfan’s Syndrome, Loeys-Dietz Syndrome, aneurysms and dissections of thoracic aorta: ACTA2, CBS, COL3A1, COL5A1, FBN1, FBN2, FLNA, MED12, MYH11, MYLK, NOTCH1, PRKG1, SKI, SLC2A10, SMAD3, SMAD4, TGFB2, TGFBR1, TGFBR2. The employed technique was Next Generation Sequencing (NGS) and validation of the pathogenetic variants through automated sequencing according to the Sanger method (Tipu et al. 2015). Research of mutations of the genes included in the panel was carried out through automated sequencing according to the Sanger method. This research was made by means of a set of primers (Ion AmpliSeqTM Cardiovascular Research Panel) producing 1043 amplicons. Analysis was performed according to the protocol of manufacturer using the Ion-Torrent PGM platform. This technique is based on sequencing by synthesis and detection trough semiconductor chip method. Coverage >20; Quality Control (QC) >20 are the criteria used for calling the variants. The presence of surely pathogenetic ones was confirmed by means of PCR amplification and traditional sequencing in accord to the Sanger method, using an automatic fluorescent technology analyser.
The NGS technology can lead to errors at different stages of the process, and it has got a sensitivity equal to 95% (Qin 2019).
The corpse belonged to a female individual assessed to be 35 years old at the time of her death. The total length of the corpse measured on the autoptic table was 185 cm. The colour of the iris was brown and a subluxation of the crystalline lens emerged from the analysis of the medical record. Inspection of the oral cavity showed no abnormalities worthy of note and the absence of dental prostheses. The examined body appeared to be characterised by regular nutrition and showed muscular trophism. The panniculus adiposus was regularly present. Striae gravidarum (stretch marks) were noted. Normally formed external genitalia were recorded.
Upper limbs = 97 cm (measuring the distance between the humeral head and the distal phalanx of the middle finger of the hand).
Lower limbs = 97 cm (measuring the distance between the anterior superior iliac spine and the sole of the foot, medially, at the level of the tuberosity of the scaphoid bone).
Right wrist circumference =17 cm.
Left wrist circumference = 17 cm.
Left ankle circumference = 23 cm.
Right ankle circumference = 23 cm.
During the dissection of the cadaver, only the presence of a dissecting aneurysm of the aorta, from the intrapericardial ascending trait to the aorto-iliac bifurcation of this artery was revealed. According to the Stanford classification, this dissection was catalogued as type A, with generation of a false lumen. At sections of the aortic lumen, both real and false one, the medium adventitial dissection was 45 cm long. The section of the intrapericardial anterior wall of the aortic root disclosed the presence of an ulcerated atheromatous plaque with a contextual intramural blood collection and a haemorrhagic suffusion in the periaortic adipose tissue and subepicardial area.
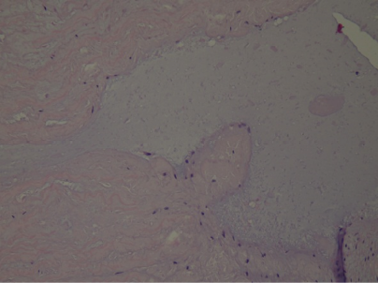
At histological examination, hematoxylin-eosin staining of the media of the aorta showed pseudocystic degeneration with deposition of mucoid substances (Figs. 1–2), and dissection of the aorta (Figs. 3, 5). Moreover, Weigert van Gieson stain evidenced disorientation and fragmentation of the elastic fibers (Fig. 4).
All these findings together allow us to postulate the presence of the Marfan’s Syndrome, as the main condition affecting the deceased patient. Specifically, regarding her cause of death, based on the evidence emerged from her autopsy, acute cardio-respiratory failure appears to be the most likely explanation. This was caused by a dissecting aneurysm of the aorta with an identified entrance in an ulcerated atheromatous plaque in the intrapericardial ascending aorta due to MFS.
Fig. 1. Histological image of intimomedial mucoid degeneration of the aorta. Hematoxylin-eosin (H/E) stain.
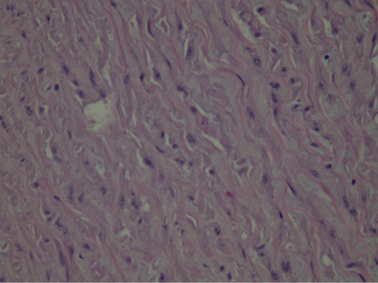
Fig. 2. Histological image of deposition of mucoid substances. H/E stain.

Fig. 3. H/E: Histological image of dissection of the aorta. H/E stain.
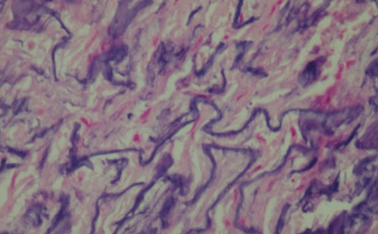
Fig. 4. W: Histological image of fragmentation of elastic fibers. H/E stain.
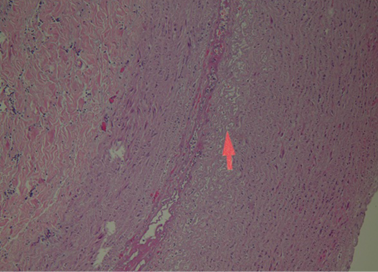
Fig. 5. H/E: Histological image of the point of least resistance in the aorta. H/E stain.
The FBN1 gene showed the presence of two de novo missense mutations: c.T6181C:p.C2061R e c.G1415A:p.C472Y.
The antiquity of MFS is currently being disputed with some maintaining that the disease can be confidently retrospectively diagnosed in ancient human remains and those who consider the provided evidence simply too scant. This applies to two cases, one a skeleton, the other a mummy respectively from the Middle Ages and the 19th century AD. In both cases, the importance of confirmatory palaeogenetic analyses has been underlined as a valuable solution to the diagnostic difficulty (Bianucci et al. 2023). Other proposals for MFS in the past have been made through the so-called “indirect” palaeopathological sources (artworks, archival and historical information) on such historical characters as Akhenaten, Paganini and Lincoln, but no conclusive evidence has so far been put forward in the available published literature (Bianucci et al. 2023).
Notwithstanding, despite its still unclarified antiquity, MFS can determine serious consequences as ventricular arrhythmias and sudden cardiac death (Hoffmann et al. 2013), for these reasons it is important a correct and multidisciplinary management of the disease. The multidisciplinary approach, as it has been illustrated above, should always take into account a potential patient’s external anatomy as well as his/her anthropometric features, although these features alone may not be sufficient to perform a definitive diagnosis and may not always allow physicians and anthropologists alike to distinguish an MFS phenotype from a marfanoid one.
In this case, analysis of the FBN1 gene showed the presence of two de novo missense mutation: c.T6181C:p.C2061R e c.G1415A:p.C472Y. These pathogenetic variants destroy a cysteine residue and so they are within the allocation criteria for de novo pathogenetic variants. To the best of our knowledge, there are no studies detailing the biological and clinical significance of both these variants. In this case, the two mutations did not cause an exuberant, externally observable marfanoid phenotype (with the exception of subluxation of the crystalline) but may reasonably have contributed to the overall pathologic habitus. Indeed, precisely thanks to the post-mortem autoptic examination, it can be asserted that the presence of fragmentation of the elastic fibers of aortic wall and myofibrils of the cardiac wall where aortic dissection is present are directly associated with the two aforementioned de novo mutations. In particular, despite from a genetic point of view they may fall into the category of the so-called Variants of Unknown Significance (VUS: variants which do not have sufficient information for attributing to them a clinical significance), it can be argued that the destruction of the cysteine residue is directly related to a significant functional mutation in FBN1.
The identified variants of FBN1, without an externally visible marfanoid phenotype, cause a fibrillin pathological dysfunction related to a predisposition to the development of aneurysms and/or aortic dissections, which may be fatal if it not promptly identified. For this reason, these two newly discovered mutations should be added to the body of knowledge on MFS.
In this preventive perspective, it is important to underline how several studies have shown favourable outcomes in pregnant patients with aortic dissection affected by MFS (Thakur et al. 2012; Allyn 2013; Chuan-Yaw et al. 2013). Surgical treatment has shown some success in type A dissection (Urbanski et al. 2013).
In conclusion, this case also shows that in subjects without symptomatic or externally evident MFS prevention is indeed crucial in order to:
Therefore, the discovery of these two de novo new mutations, provided by an autoptic and forensic anthropological study, could also improve the effectiveness of genetic screening, especially in patients who might not be straightforwardly classified as MFS sufferers based on gross clinical examinations (Aquila et al. 2020).
This case also shows how a confirmatory diagnosis of MFS requires the interplay of medicine (clinical and forensic), the morpho-anthropological sciences and genetics in order to attain a more precise understanding of the phenotype and genotype of this still mysterious condition, a challenge both to physicians and anthropologists.
Conflict of interest statement
The authors have no conflict of interest to report.
Authors’ contributions
I.A. – conceptualisation, writing of the first draft, diagnostic workflow; data acquisition and analysis; I.A., M.A.S., S.B., D.M., G.V., L.A., M.M., E.V., F.M.G., P.R. – critical revision of the manuscript, editing, revision of the first draft and response to the reviewers’ comments, data analysis, data acquisition; P.R. – senior supervision.
Allyn J, Guglielminotti J, Omnes S, Guezouli L, Egan M, Jondeau G, et al. 2013. Marfan’s syndrome during pregnancy: anesthetic management of delivery in 16 consecutive patients. Anesth Analg 116(2):392–8. https://doi.org/10.1213/ANE.0b013e3182768f78
Asgari S, Luo Y, Akbari A, Belbin GM, Li X, Harris DN, et al. 2020. A positively selected FBN1 missense variant reduces height in Peruvian individuals. Nature 582(7811):234–9. https://doi.org/10.1038/s41586-020-2302-0
Aquila I, Sacco MA, Cordasco F, Ricci P. 2020. Forensic case of a pregnant woman with Marfan syndrome. BMJ Case Rep 13(12):e229959. http://doi.org/10.1136/bcr-2019-229959
Bianucci R, Donell S, Galassi FM, Lanza T, Mattutino G, Nerlich A, Sineo L. 2023. Marfan Syndrome in Palaeopathology: A review. Hum Evol 38(1–2):29–36. https://doi.org/10.14673/HE2023121111
Chuan-Yaw C, Jean-Ming Y, Chon-Wa L, Pi-Hua C. 2013. Successful management of aortic dissection in a patient with Marfan syndrome during pregnancy. Am J Obstet Gynecol 208(2):e3–6. https://doi.org/10.1016/j.ajog.2012.11.034
Dean JCS. 2007. Marfan syndrome: clinical diagnosis and management. Eur J Hum Genet 15:724–33. https://doi.org/10.1038/sj.ejhg.5201851
Dolci C, Pucciarelli V, Gibelli DM, Codari M, Marelli S, Trifirò G, Pini A, Sforza C. 2018. The face in Marfan syndrome: A 3D quantitative approach for a better definition of dysmorphic features. Clin Anat 31(3):380–6. https://doi.org/10.1002/ca.23034
Frydman M. 2008. The Marfan Syndrome. Isr Med Assoc J 10(3):175–8.
Hoffmann BA, Rybczynski M, Rostock T, Servatius H, Drewitz I, Steven D et al. 2013. Prospective risk stratification of sudden cardiac death in Marfan’s syndrome. Int J Cardiol 167(6):2539–45. https://doi.org/10.1016/j.ijcard.2012.06.036
Jensen SA, Atwa O, Handford PA. 2021. Assembly assay identifies a critical region of human fibrillin-1 required for 10-12 nm diameter microfibril biogenesis. PLoS One 16(3):e0248532. https://doi.org/10.1371/journal.pone.0248532
Klintschar M, Bilkenroth U, Arslan-Kirchner M, Schmidtke J, Stiller D. 2009. Marfan syndrome: clinical consequences resulting from a medicolegal autopsy of a case of sudden death due to aortic rupture. Int J Legal Med 123(1):55–8. https://doi.org/10.1007/s00414-008-0288-5
Krause KJ. 2000. Marfan syndrome: literature review of mortality studies. J Insur Med 32(2):79–88.
Dietz HC. Marfan Syndrome. 2001 Apr 18 [Updated 2011 Dec 1]. In: Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews™[Internet]. Seattle (WA): University of Washington, Seattle; 1993. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1335/
Qin D. Next-generation sequencing and its clinical application. 2019. Cancer Biol Med 16(1):4–10. https://doi.org/10.20892/j.issn.2095-3941.2018.0055
Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103:2499–509. https://doi.org/10.1083/jcb.103.6.2499
Thakur V, Rankin KN, Hartling L, Mackie AS. 2012. A systematic review of the pharmacological management of aortic root dilation in Marfan syndrome. Cardiol Young 23(4):568–81. https://doi.org/10.1017/S1047951112001412
Tipu HN, Shabbir A. 2015. Evolution of DNA sequencing. J Coll Physicians Surg Pak 25(3):210–5.
Trawicka A, Lewandowska-Walter A, Majkowicz M, Sabiniewicz R, Woźniak-Mielczarek L. 2022. Health-Related Quality of Life of Patients with Marfan Syndrome-Polish Study. Int J Environ Res Public Health 19(11):6827. https://doi.org/10.3390/ijerph19116827
Urbanski PP, Hijazi H, Dinstak W, Diegeler A. 2013. Valve-sparing aortic root repair in acute type A dissection: how many sinuses have to be repaired for curative surgery? Eur J Cardiothorac Surg 44(3):439–43; discussion 443–4. https://doi.org/10.1093/ejcts/ezt042
Wozniak-Mielczarek L, Osowicka M, Radtke-Lysek A, Drezek-Nojowicz M, Gilis-Malinowska N, Sabiniewicz A et al. 2022. How to Distinguish Marfan Syndrome from Marfanoid Habitus in a Physical Examination-Comparison of External Features in Patients with Marfan Syndrome and Marfanoid Habitus. Int J Environ Res Public Health 19(2):772. https://doi.org/10.3390/ijerph19020772

