
Available online at: https://doi.org/10.18778/1898-6773.85.2.03
Universidad Autónoma de Madrid. Departamento de Biología. Unidad de Antropología Física, Madrid, Spain
ORCID: 0000-0002-5175-4385
Sección de Antropología Forense, Instituto de Medicina Legal de Madrid, Madrid, Spain
ORCID: 0000-0002-1137-2016
FAPAB Research Center, Avola (SR), Sicily, Italy
Archaeology, College of Humanities, Arts and Social Sciences, Flinders University, Adelaide, SA, Australia
Department of Anthropology, Faculty of Biology and Environmental Protection, University of Lodz, Lodz, Poland
ORCID: 0000-0001-8902-3142
FAPAB Research Center, Avola (SR), Sicily, Italy
Archaeology, College of Humanities, Arts and Social Sciences, Flinders University, Adelaide, SA, Australia
ORCID: 0000-0001- 6637-9402
Hospital General de Catalunya, Spain
ABSTRACT: The aim of this study is to show the cranial alterations that Klippel-Feil syndrome produced in a case older than 200 years. Few paleopathological case studies diagnosed as Klippel-Feil Syndrome are focused on cranial abnormalities. A skull numbered 778, belonging to the Federico Olóriz Aguilera collection (Spain, 19th century AD), Universidad Complutense de Madrid, belonging to a young man born in a town in the North of Spain, was investigated. This cranium was visually inspected, hence macroscopically and paleoradiologically studied, using the images obtained through conventional radiology and CT scan imaging. In addition to the vertebral fusion between the atlas (C1) and the axis (C2), atlanto-occipital fusion, basilar impression, obliteration of the sagittal suture, enlarged parietal foramina and significant craniofacial asymmetry affecting maxillary bones, sphenoid, orbits, nasal bones and both palatines were observed. Morphological findings make it possible to diagnose a Klippel-Feil syndrome, possibly type-II, although the lack of the rest of the spinal column renders it impossible to verify other spinal anomalies. As a limitation, only the cranium and two cervical vertebrae were preserved, hence the possible involvement of the rest of the skeleton cannot be verified.
KEY WORDS: Olóriz collection, Klippel-Feil syndrome, atlanto-occipital fusion, basilar impression, facial asymmetry, enlarged parietal foramina
Klippel-Feil syndrome (KFS) is characterized by the congenital fusion of a variable number of cervical vertebrae (Vujasinovic Stupar et al. 2015), in the majority of cases accompanied by other abnormalities in different apparatuses, including otorhinolaryngological and craniofacial ones (Clarke et al. 1998). The classic clinical KFS triad comprises a short neck, restricted neck mobility, and a low dorsal hairline (Gunderson et al. 1967; Fietti and Fielding 1976; Taylor-Martínez et al. 2019). In the modern clinical setting, in cases in which the condition presents asymptomatically it can be discovered by chance, after a radiological examination in living patients. Although the etiology of KFS is not fully understood, some genetic involvement was postulated since mutations in the GDF6, GDF ٣, and MEOX1 genes were found in some families and an inheritance pattern was determined. For the first two, involved in bone formation and development, it is an autosomal dominant pattern, while for the third one, whose homeobox protein regulates vertebral separation, it is instead an autosomal recessive one (Manger et al. 2021). Before genetic correlations were found, families with KFS individuals within them were historically subjected to anthropological studies (Henneberg and Otocki 1974).
Several cases of KFS have been recorded in the paleopathological literature. Archaeological cases globally have been described since the Neolithic in Slovakia (site of Vráble-Veľké Lehemby) and Late-Final Neolithic in Greece (Alepotrypa Cave, Peloponnese, 5000-2300 BC) (Papathanasiou 2005; Hukeľová et al. 2021), the Chalcolithic Age in Peru (MacCurdy 1923), North America (Jarcho 1965), Central America (Urunuela and Alvarez 1994), Ancient Egypt (Aufderheide and Rodriguez-Martin 1998) or Europe (Barnes 1994; Gladykowska-Rzeczycka 1997; González-Reimers et al. 2001; Herrerín, 2004; 2011; Pany and Teschler-Nicola 2007; Fernandes and Costa 2007; Giuffra et al. 2009; Macías-López 2020). In Italy, four cases of KFS from the 1582–1583 AD plague phase of the San Michele cemetery in Alghero (Sardinia) were identified among 199 individuals making this the archaeological site with more such finds also dating to the same historical period (Varotto et al. 2020).
In this study the case of a skull from the Olóriz collection is presented: this specimen shows some of the typical features of KFS, despite the sole preservation of two cervical vertebrae and not of the entire skeleton. The Olóriz collection was formed for research purposes – as the history of anatomy teaches (Papa et al. 2020) – from corpses collected by Federico Olóriz Aguilera (1855–1912), Professor of Anatomy at the University of Madrid, reaching a total of 2,250 skulls, almost entirely from Spain and dating to the 19th century. At present the collection is distributed among various Departments of the Universidad Complutense de Madrid, and although the written documentation is now missing, all the skulls are marked with black ink indicating their basic data about sex, age at death and geographic provenance. In addition, it must be underlined that the collection is dispersed and many specimens have disappeared from the register. However, it has been possible to verify that there is no similar case among those preserved, after a thorough review of all the preserved skulls.
The analyzed skull (inventory number 778 – Olóriz collection) comes from Viana del Bollo, in the Spanish province of Orense. Data about sex and age at death are labelled on the skull itself, but sex determination and age estimation were anyhow performed utilizing Ferembach et al.’s (1979), Masset’s (1982) and Buikstra and Ubelaker’s (1994) sets of methods. The skull was completely preserved, although the mandible is missing, and, as a result on the fusion, the atlas and axis are still present. Its state of preservation can be grossly assessed as good although no preservation indexes, more commonly used for bioarchaeological material originating from excavation sites – and, as yet, not for scientifically musealized anatomical specimens – were calculated. Together with the visual inspection and macroscopic examination of the skull, image analysis was carried out using conventional radiology and CT scan imaging (Equipment: HP 15.0; Parameters: 120kV/150mAs; 0.5/3.0mm).
The information about sex and age at death was written on the right parietal bone by Dr. Olóriz himself (Fig. 1): the skull belongs to a 22-year-old male. Morphologically and anthropologically, the skull confirmed the above-stated information: male features and 20–25 years age range.
The following pathological findings were observed in skull 778.
Neurocranium:
Splanchnocranium:
Spinal column:
Fig. 1. Lateral view of skull number 778 with information on sex, age at death and provenance labelled on the ectocranial surface of the right parietal bone.
In the superior view (Fig. 2a), the sagittal suture was totally missing and the coronal one showed no obliterated areas. In the frontal view, a significant lateral asymmetry was observed, with deviation of the nasal bones and nasal septum to the right (Fig. 2b). This asymmetry also affected the orbits, the left being more cranially located, taller and narrower than the right. In the posterior view (Fig. 2c), the asymmetry was also evident, with significant lateral displacement of the sagittal suture and the lambda craniometric point, while the lambdoid suture appeared visible and unobliterated. Additionally, two parietal enlarged foramina are present (Fig. 2c).
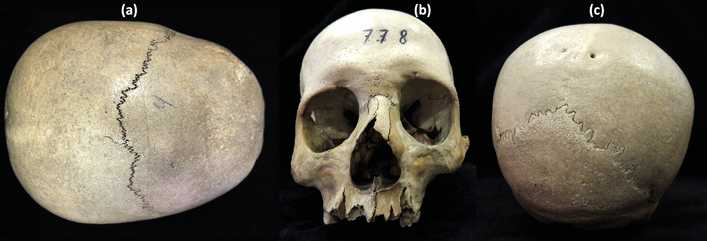
Fig. 2. Macroscopical examination. a) Superior view; b) Frontal view; c) Posterior view.
In the inferior view the skull showed asymmetry of the entire facial massif (Fig. 3a, b), with a very significant displacement of the median sagittal plane (red and white lines). This asymmetry was very evident in the maxillary palatal processes and palatal bones, with smaller dimensions in the left hemicranium (Fig. 3c).
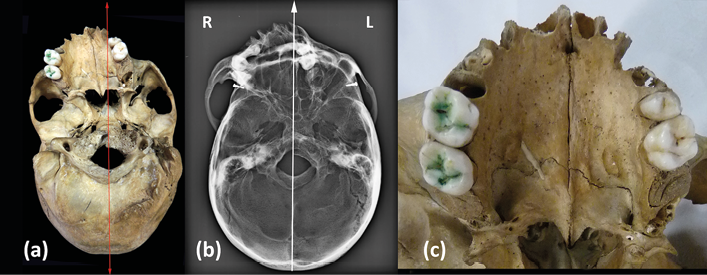
Fig. 3. a) Inferior view (red line, sagittal plane); b) Inferior view, X-Ray image (white line, sagittal plane);
c) Maxillar and palatal bones, inferior view.
The palatal suture was asymmetrical in its course and length. Four teeth remained in situ (16, 17, 25 and 26) as well as two roots (18, 27), while the rest were lost post-mortem. The postero-inferior view (Fig. 4a) showed an atlanto-occipital fusion, with fusion of the lateral massif and a normal appearing posterior arch of the atlas. The axis was found to be fused with the atlas at its articular facets. The odontoid process was fused with the anterior arch and the right lateral mass of the atlas. Part of the odontoid process had already been lost historically, torn off as a result of past manipulations performed to separate the skull from the spine at the time of its musealization (Fig. 4a+). On the other hand, the atlas was fused to the occipital but not symmetrically, leaving a space between the left postero-lateral edge of the foramen magnum and the left postero-lateral arch of the atlas (Fig. 4a*). The lower view (Fig. 4b) also showed the reduction in the antero-posterior diameter of the vertebral canal.
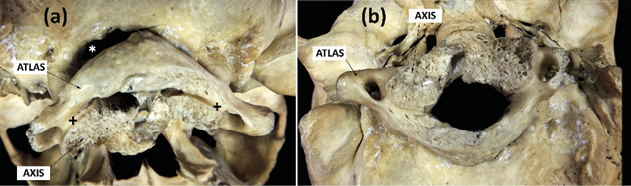
Fig. 4. Postero-inferior view. a) atlanto-occipital fusion + axis-atlas fusion. * Space between the left postero-lateral edge of the foramen magnum and the left postero-lateral arch of the atlas. b) Stenosis of the vertebral canal.
Conventional radiographic analysis showed a 4 mm basilar impression on McGregor’s line (hard palate-occipital scale; red line; Fig. 5a). At the same time the CT study made it possible to verify how the asymmetry also affected the sphenoid bone, with a marked shift towards the right side of its pterygoid processes (Fig. 5b), and likewise the maxillary bone and the nasal cavity (Fig. 5c). Finally, the tomography verified the stenosis of the vertebral canal at the level of the foramen magnum (Fig. 5d, e), due to the fusion of the atlas in an asymmetric, rotated position.
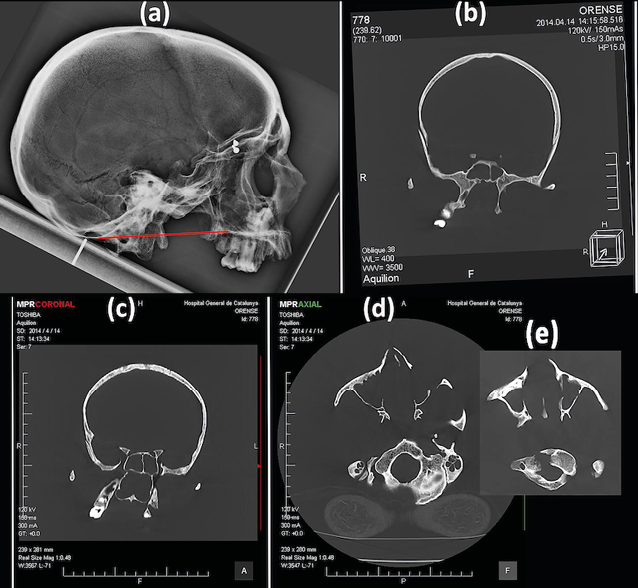
Fig. 5. a) Conventional radiographic image. McGregor’s line (red line). b) CT image. Frontal view. Deviation of the pterygoid processes. c) CT image. Frontal view. Deviation of the maxillary bone and the nasal cavity d) and e) CT image. Inferior view. Stenosis of the vertebral canal at the level of the foramen magnum.
KFS was first described in 1912 by Maurice Klippel and André Feil (1912) in a 46-year-old patient with a massive fusion of the cervical vertebrae, although the first clinical descriptions are attributed in 1745 to Haller and in 1746 to Morgagni (Gunderson et al. 1967).
Three different forms of KFS are defined (Barnes 1994; Pany and Teschler-Nicola 2007; Toker et al. 2009):
Therefore, the cervical vertebrae are the most commonly affected, leading to the description, especially in old clinical textbooks, of a syndrome in which patients are characterized by a shortened or absent neck as the result of a complete cervical block.
The prevalence of KFS is estimated at 0.71% (Brown et al. 1964; Papagrigorakis et al. 2003; Nouri et al. 2017) and the incidence of one case per 30,000–42,000 (de Rubens-Figueroa et al. 2005), with a female predilection (Jones 1997; Aufderheide and Rodriguez-Martin 1998; Martínez-Quintana and Rodríguez-González 2015; Sirico et al. 2015; Gruber et al. 2018).
The final cause of this vertebral fusion is found in a failure in spinal segmentation between the third and eighth weeks of embryogenesis (Fietti and Fielding 1976; Mahirogullari et al. 2006; Fernandes and Costa 2007). There is no unanimity of criteria on its genetic origin (Daum and Jones 1988; de Rubens-Figueroa et al. 2005; Pany and Teschler-Nicola 2007; Toker et al. 2009), with a genetic inheritance being proposed, dominant in Type II and recessive in Types I and III (Gunderson et al. 1967; Juberg and Gershanik 1976; Lowry et al. 2001).
Cervical fusions in KFS can remain asymptomatic, it being discovered accidentally after radiological analysis (Copley and Dormans 1998), although limitation of neck movement and its relationship with other abnormalities occurs in a high percentage of cases (Dietz 2001). On the other hand, atlanto-occipital fusion can lead to severe neurological symptoms after even minor trauma (Gray et al. 1964) due to the excessive mobility of the vertebral segments adjacent to the fused area (Strax TE, Baran E. 1975; Adeleye and Akinyemi 2010).
Multiple disorders associated with KFS have been described, such as kyphosis, spina bifida, cleft palate, atlanto-occipital fusion, basilar impression, scoliosis, supernumerary cervical vertebra, Sprengel’s deformity, dwarfism, hypodontia, meningocele, renal and cardiac anomalies (Gunderson et al. 1967; Hensinger et al. 1974; Hensinger and MacEwen 1986; Daum and Jones 1988; Tachdijian 1990; Barnes 1994; Copley and Dormans 1998; Herman and Pizzutillo 1999; Warner 1998; Dietz 2001; Papagrigorakis et al. 2003; Narang and Goyal 2006; Pany and Teschler-Nicola 2007; Toker et al. 2009). Specifically, atlanto-occipital fusion is produced by a failure in the segmentation between the skull and the first cervical vertebra. It can be partial or complete, and it usually produces associated basilar impression (Boleaga-Durán et al. 2006). It can appear as an isolated sign or be part of different syndromes, among them (very frequently), type-II Klippel-Feil syndrome. With particular reference to the basilar impression or basilar invagination, it consists of an elevation of the floor of the posterior fossa, with displacement of the odontoid process towards the interior of the foramen magnum (Chamberlain 1939). The primary or malformative basilar impression is almost always associated with atlanto-occipital fusion and narrowing of the foramen magnum (List 1941). Among the many causes that can produce a basilar impression (osteomalacia, Paget’s disease, Chiari malformation, syringomyelia, hydrocephalus, etc.), the Klippel-Feil syndrome is counted (Matson 1969). In skull 778, the radiographic analysis showed a light but obvious basilar impression (4 mm) on McGregor’s line (hard palate-occipital scale; red line; Fig. 5a). Moreover, two enlarged parietal foramina (EPF) detected in the parietal bones are present. EPF are developmental defects characterized by variable intramembranous ossification, normally located on each side of the dorsal portion of the sagittal suture. They differ from normal parietal foramina, which are smaller (less than 1 mm in diameter) and considered anatomical variants. EPF can be associated with syndromic condition such as KFS or Saethre-Chotzen syndrome (Thompson et al. 1984) or can be found isolated (Piagkou et al. 2013).
A typical paleopathological diagnosis of KFS involves a morphological examination of the column and any other areas of the skeleton to determine if other anomalies may be present, a paleopathological and clinical comparison with the available scientific literature, and an imaging study (conventional X-ray and/or CT scan) in order to confirm the suspected skeletal condition ruling out other pathologies such as fusion caused by traumatic conditions like fractures. Occasionally, but not routinely (especially with historic remains in which invasive sampling is not always justified), a genetic test can be made to corroborate phenotypic observations with the discovery of a matching genotypic background. A sample may be taken and examined to see if there are mutations in genes like GDF3, GDF6, or MEOX1, which are known to be commonly affected in KFS (Mohamed et al. 2013). However, it must be stressed that, both clinically and paleopathologically, radiological analyses can be considered sufficient to make a substantiated diagnosis of KFS.
Differential diagnoses include Paget’s disease, fibrous dysplasia, cleidocranial dysostosis, osteogenesis imperfecta, osteoporosis, rickets.
Paget’s disease: Paget’s disease of bone is a chronic bone disorder of unknown cause. It was first described by Sir James Paget (1814–1899) in 1877. There is an increase in osteoclast activity, resulting in increased bone resorption, the clinical expression of which is the lytic bone lesions observed in conventional radiography (Resnik and Niwayama 1988; Bolland and Cundy 2013). In response, accelerated and chaotic bone formation occurs, resulting in sclerotic bone that is functionally weaker than normal bone without the characteristic laminar pattern (Menéndez-Bueyes and Soler-Fernández 2017). The clinical manifestations are usually expressed after years of evolution when bone deformity appears that leads to pain, osteoarthritis, and pathological fractures (Roodman and Windle 2005; Bolland and Cundy 2013; Corral-Gudino et al. 2013; Galson and Roodman 2014). The disease usually presents at an older age than 55 and its frequency increases with age (Resnik and Niwayama 1988; Bolland and Cundy 2013), with a slight predominance in males. The bones most frequently involved are the pelvis, femur, spine, skull, and tibia (Bolland and Cundy 2013). In the postcranial skeleton, thickening of the diaphyses of the limb bones are observed, which tend to deform laterally and fracture (in extreme affectations), with the femur and tibia being the most affected bones (Ortner 2003). The alteration in the cranium can cause a symmetrical or asymmetric growth of the parietal or frontal bones causing a greater size of the cephalic portion (Favus and Vokes 2005). This cranial expansion can narrow the diameter of the cranial foramina and cause neurological complications including hearing loss due to cochlear nerve damage caused by involvement of the temporal bone, cranial nerve palsy, and softening of the skull base with risk of compression of the brainstem. In the facial bones, a deformity and/or loss of teeth is caused (Aufderheide and Rodríguez-Martín 1998; Ortner 2003; Favus and Vokes 2005).
Of all the listed features, only platybasia is present in skull 778. But some features of skull 778 may allow this disease to be ruled out, even though the vertebral column and the rest of the postcranial skeleton have not been preserved. First of all, age makes it possible to exclude this possibility. It would be extremely rare for a 22-year-old young adult to show signs of Paget’s disease as advanced as the pathological changes shown in this case. Secondly, the widening of the diploe (hyperostosis of the cranial vault), the typical “cottony” radiological image of Paget’s disease and circumscribed osteoporosis, do not appear in this case (Resnick and Kransdorf 2006; Herrerín et al. 2009). Third, CT images do not show involvement of the temporal bones, or stenosis of the auditory canal. And, lastly, teeth are not affected.
Fibrous dysplasia (FD): it is a bone development disorder in which the lesions form fibrous tissue and spicules of bone tissue (Ortner 2003). The spongy medullary bone is replaced by fibrous tissue. It can be monostotic (a single affected bone) or polyostotic (multiple lesions). The monostotic form is the most common (Herrerín et al. 2009). The monostotic forms mainly affect the long bones, ribs and radius. The polyostotic forms usually involve the proximal femur and the base of the skull (Parekh et al. 2004; Alonso and Muñoz-Torres 2009). The most frequently affected bone in fibrous dysplasia is the femur (44%) followed by the skull (38%), the pelvis (23%), the ribs (16%) and the spine (9%) (Benhamou et al., 2006). When the skull is affected, there is an expansion of the diploe associated with a reduction in the thickness of the internal and external tables of the cranial vault (Herrerín et al. 2009). Radiographic images are very similar to those obtained in Paget cases (cottony images). But computed tomography is more important in diagnosis, because it shows expansive focal areas with a homogeneous “ground glass” appearance (Herrerín et al. 2009; Raus and Coroiu 2016). Despite not having recovered the postcranial skeleton, we ruled out fibrous dysplasia because it does not show a widening of the diploe or the typical CT image in “thin glass” (Herrerín et al. 2009).
Cleidocranial dysostosis (CCD). The pathogenesis of CCD is currently unknown: it is probably caused by an ectodermal and mesodermal tissue disorder during the bone growth phase (Hernández et al., 1980). The difficulty in making a diagnosis of CCD lies in the variability of the alterations. The skull has the anthropometric characteristics of brachycephaly, protruding fronto-parietal fontanelle with large sutures and numerous small supernumerary (Wormian) bones (Herrerín 2011). Affected individuals show a slight hypertelorism and exophthalmos (Hernández et al., 1980; Ortner, 2003; Roberts et al., 2013; Russell, 2015; Lewis, 2019). In the dentition, skull 778 does not show supernumerary teeth or agenesis of premolars, which are also frequently seen signs in patients with CCD (Herrerín 2004; 2011).
Osteogenesis imperfecta (OI): OI are a group of inherited genetic pathologies of the connective tissue characterized by bone fragility and fractures (Jones 2006). They result from constitutional bone fragility (cortical bone thinning, trabecular bone rarefaction) but also from acquired bone fragility due to muscle wasting and immobilization. Wide fontanelles are a known symptom of OI. Typical radiographic signs are thinning of the cortical bone and excessive transparency of the trabecular bone. The main radiographic features are osteopenia, bone fractures and bone deformities (Renaud et al. 2013). These signs do not appear on this skull.
Osteoporosis: there are no radiological osteopenic signs suggesting some form of osteoporosis (Resnick and Kransdorf 2006). The age of the individual (22 years) also makes this disease very unlikely.
Rickets: rickets is ruled out in the absence of cranial thickening and cranial porosity, frequent in the frontal and parietal bones of individuals affected by this disease (Resnick and Kransdorf 2006).
Among the various craniofacial anomalies associated with KFS, facial asymmetry occurs in 13% to 20% of cases (Martínez-Quintana and Rodríguez-González 2015; Naikmasur et al. 2011; Kerai and Saxena 2014; Jovankovičová et al. 2012). This facial asymmetry is very significant in skull 778, including the nasal area, where the part of piriform aperture and the nasal bones are asymmetrical, with the nasal septum laterally deviated from the sagittal plane. Both the displacement of the maxillary mass and the asymmetry of the orbits and nasal region may have their origin in genetic factors (as occurs in the case of hemifacial microsomia, multiple syndromes, craniosynostosis of the coronal suture or labio-palatal fissures) or in environmental and/or functional factors (such as intrauterine pressure, especially in multiple births, ante-mortem trauma with deficient fusion or even infections in the area during growth). In this skull we have not found any sign that would allow us to relate it to synostosis of the coronal suture, as this is the only synostosis that can present asymmetrical compensation of the cranial and facial bones. Neither have we detected, both in the conventional radiological study and in the CT scan, the presence of signs of any ante-mortem fracture nor any signs of infection.
These nasal features have also been described in other patients with KFS (Fragoso et al. 1982). The asymmetry of the palatine processes of the maxilla and palatine bones observed are also not unusual findings in KFS cases with facial asymmetry (Martínez-Quintana and Rodríguez-González 2015). Giuffra et al. (2009) describe the case of Cardinal Carlo de’ Medici (1595–1666), who had a clear asymmetry of the nasal and maxillary bones, larger on the left side, together with a marked hypoplasia of the right hemimandible. All of these characters produced an easily recognizable facial asymmetry in the portrait that the artist Justus Sustermans (Galleria Palatina, Rome) made of the Cardinal.
Anomalies associated with KFS, such as atlanto-occipital fusion or basilar impression are also rarely mentioned in the paleopathological literature. The fusion of the atlas has been described in an individual of medieval chronology, in Portugal (13th–15th centuries), presenting partial atlanto-occipital fusion together in the skull with very slight differences in the outline of the nasal cavity (Fernandes and Costa 2007). Regarding the atlanto-occipital fusion, we have not found signs of infection, fracture or rheumatoid arthritis (subluxation, erosion, sclerosis, basilar impaction, etc.), which could indicate a different origin than congenital. In a juvenile individual from Gnadendorf (Hungary, 10th century AD), in addition to congenital fusion of several cervical and thoracic vertebrae, symmetric hypoplasia of the occipital bone, marked curvature of the occipital scale, basilar impression, and asymmetry of the occipital condyles have been described (Pany and Teschler-Nicola 2007). Such malformations are rarely described in the anthropological literature. In the presented specimen, none of these signs are present. The age of the individual (22 years), together with the data provided by the analysis of the radiological images, has made it possible to rule out other cranial malformations with similar signs, such as Paget’s disease, fibrous dysplasia, cleidocranial dysostosis, osteogenesis imperfecta, osteoporosis or rickets. Therefore, in skull 778, all the described findings are compatible with Type-II KFS, although, as only two cervical vertebrae are available, a completely accurate diagnostic assessment cannot be made. Type-II KFS corresponds to about 26% of all cases of this syndrome (McGaughran 2004), and usually includes atlanto-occipital fusion.
The pathological findings observed in skull 778 include the absence of sagittal suture, enlarged parietal foramina, basilar impression, atlanto-occipital fusion, asymmetry of orbits and nasal region, asymmetry of nasal and palatal bones, displacement of maxillary massif and C1-C2 fusion. This leads to the diagnosis of Type-II Klippel-Feil syndrome. X-rays and CT scans corroborate the morphological findings. Due to the absence of most of the vertebral column, in this article we have also paid a great of attention to other important skeletal alterations at the cranial level which are known to accompany this syndrome and, in this particular, have its detection more straightforward. This description also adds to the list, still under scrutiny, of previously reported KFS cases in the Spanish bioarchaeological record, covering a chronological span from the Bronze Age to the 19th century AD. As a final general note to this historical dissertation, it must be stressed how a careful analysis of cranial asymmetries and splanchnocranial modifications should always constitute an important part of retrospective assessments of this condition.
Acknowlegdments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors contribution
J.H., E.D., R.D.S. = conceptualization, first draft, writing, data analysis and synthesis, diagnosis, palaeoradiological analysis;
F.M.G, E.V = writing, data analysis and synthesis, diagnosis, palaeoradiological analysis, literature review
Conflict of interest
The authors declare they have no conflict of interest.
Adeleye AO, Akinyemi RO. 2010. Cervical Klippel-Feil syndrome predisposing an elderly African man to central cord myelopathy following minor trauma. Afr Health Sci 10:302–4.
Alonso G, Muñoz-Torres M. 2009. Displasia ósea fibrosa en un varón joven. Endocrinol Nutr 56:195–200.
Aufderheide AC, Rodríguez-Martín CR. 1998. The Cambridge Encyclopedia of Human Paleopathology. Cambridge: Cambridge University Press.
Barnes E. 1994. Developmental Defects of the Axial Skeleton in Paleopathology. Colorado: University Press of Colorado.
Benhamou J, Gensburger D, Messiaen C, Chapurlat R. 2016. Prognostic factors from and epidemiologic evaluation of fibrous dysplasia of bone in a modern cohort: the FRANCEDYS Study. J Bone and Miner Res 12:2167–72.
Boleaga-Durán B, Suárez E, Tomasini Ortiz P, Reyes J. 2006. Anatomía y patología de la unión craneovertebral. Anales de Radiología México 2:153–70.
Bolland MJ, Cundy T. 2013. Paget’s disease of bone: Clinical review and update. J Clin Pathol 66:924–7.
Brown MW, Templeton AW, Hodges FJ. 1964. The incidence of acquired and congenital fusions of the cervical spine. Am J Roentgenol Radium Ther Nucl Med 92:1255–9.
Buikstra JE, Ubelaker DH. 1994. Standards for data collection from human skeletal remains. Fayetteville: Arkansas Archeological Survey Research Series, No. 44.
Chamberlain WE. 1939. Basilar impression (platybasia). A bizarre developmental anomaly of the occipital bone and upper cervical spine with striking and misleding neurologic manifectations. Yale J Biol Med 11:487–96.
Clarke RA, Catalan G, Diwan AD. 1998. Heterogeneity in Klippel-Feil syndrome: a new classification. Pediatr Radiology 28:967–74.
Copley LA, Dormans JP. 1998. Cervical spine disorders in infants and children. J Am Acad Orthop Surg 6:204–14.
Corral-Gudino L, Borao-Cengotita M, Pino-Montes J, Ralston S. 2013. Epidemiology of Paget’s disease of bone: A systematic review and meta-analysis of secular changes. Bone 55:347–52.
Daum REO, Jones DJ. 1998. Fiberoptic intubation in Klippel-Feil syndrome. Anaesthesia 43:18–21.
de Rubens-Figueroa J, Zepeda-Orozco G, González-Rosas A. 2005. Síndrome de Klippel-Feil: una enfermedad musculoesquelética, con malformaciones cardiovasculares asociadas. Bol Med Hosp Infant Mex 62:348–55.
Dietz F. 2001. Congenital Abnormalities of the Cervical Spine. In: Weinstein SL, editor. The Pediatric Spine Principles and Practice, Second Edition. Philadelphia: Lippincott Williams & Wilkins, 239–51.
Favus M, Vokes T. 2005. Paget disease and other dysplasias of the bone. In: Kasper DL et al., editors. Harrison’s Principles of Internal Medicine. 16th edition. New York: McGraw-Hill, 2283–4.
Ferembach D, Schwidetzky I, Stloukal M. 1979. Recommandations pour determiner l’age et le sexe sur le squelette. Bull et Mèm. de la Soc. d’Anthrop. de Paris 6 série XIII: 7–45.
Fernandes T, Costa C. 2007. Klippel–Feil syndrome with other associated anomalies in a medieval Portuguese skeleton (13th–15th century). J Anat 211:681–5.
Fietti VG, Fielding JW. 1976. The Klippel-Feil syndrome: Early roentgenographic appearance and progression of the deformity. J Bone Joint Surg 58A:891–2.
Fragoso R, Cid-García A, Hernández A, Nazará Z, Cantú JM. 1982. Frontonasal dysplasia in the Klippel-Feil syndrome: a new associated malformation. Clin Genet 22: 270–3.
Galson DL, Roodman GD. 2014. Pathobiology of Paget’s disease of bone. J Bone Metab 21:85–98.
Giuffra V, Vitiello A, Giusiani S, Fornaciari A, Caramella D, Villari N, Fornaciari G. Rheumatoid arthritis, Klippel-Feil syndrome and Pott’s disease in Cardinal Carlo de’ Medici (1595–1666). 2009. Clin Exp Rheumatol 27:594–602.
Gladykowska-Rzeczycka J. 1997. A serious defect of two cervical vertebrae from a medieval cemetery in Poland; Klippel-Feil syndrome? Acta Biol Szeged 42:49–53.
González-Reimers E, Mas-Pascual A, Arnay-de-LaRosa M, Velasco-Vázquez J, Jimenéz-Gómez MC. 2001. Klippel-Feil syndrome in the prehispanic population of El Hierro (Canary Islands). Ann Rheum Dis 60: 174.
Gray SW, Romaine CB, Skandalakis JE. 1964. Congenital fusion of the cervical vertebrae. Surg Gynecol Obstet 118:373–85.
Gruber J, Saleh A, Bakhsh W, Rubery PT, Mesfin A. 2018. The prevalence of Klippel-Feil syndrome: a computed tomography-based analysis of 2,917 patients. Spine Deform 6:448–53.
Gunderson CH, Greenspan RH, Glaser GH, Lubs H. 1967. The Klippel-Feil syndrome: Genetic and clinical reevaluation of cervical fusion. Medicine (Baltimore) 46: 491–512.
Henneberg M, Otocki P. 1974. Wyniki badań antropologicznych przypadku zespołu KlippelFeila z rozszczepem podniebienia. Przegląd Antropologiczny 40:349–353.
Hensinger RN, Lang JE, MacEwen GD. 1974. Klippel-Feil syndrome: A constellation of associated anomalies. J Bone Joint Surg 56A:1246–53.
Hensinger RM, MacEwen GD. 1986. Anomalías congénitas de la Columna Vertebral. In: Rothman RH, Simeone FA (eds.), La Columna Vertebral. Madrid: Médica Panamericana, D.L. 212–344.
Herman MJ, Pizzutillo PD. 1999. Cervical spine disorders in children. Orthop Clin North Am 30:457–66.
Hernández D, Lòpez A, Paz J, Menéndez YA, Amigò YA. 1980. Enfermedad de Pierre Marie–Satnton. Incidencia familiar. Rev Esp Cir Osteoartic 15:47–57.
Herrerín J. 2004. Paleopatología. Necrópolis de El Burgo de Osma (s. XVII–XVIII). Soria, España: Soria Edita.
Herrerín J. 2011. Paleopathological discoveries in an unusual necropolis of mendicants. Soria, Spain: Soria Edita.
Herrerín J, Baxarias J, Garcia-Guixé E, Mas Pascual MA, Mariñoso ML. 2009. La anatomia patologica como factor clave en el diagnóstico de las displasias e hiperplasias craneofaciales: el error diagnóstico y radiológico. In: Polo-Cerda, M., García-Prosper, E. (Eds). Investigaciones histórico-médicas sobre salud y enfermedad en el pasado: actas del IX Congreso Nacional de Paleopatología, Morella (Castelló), 26–29 septiembre de 2007. Valencia: Grupo Paleolab & Sociedad Española de Paleopatología. 299–312.
Hukeľová Z, Krošláková M. 2021. Klippel-Feil syndrome cases from Slovakia. Int J Paleopathol 33:188–195.
Jarcho S. 1965. Anomaly of the vertebral column (Klippel-Feil Syndrome) in American Aborigines. JAMA 193:187–8.
Jones K. 2006. Smith’s Recognizable Patterns of Human Malformation. 6th Edition, Philadelphia: Elsevier Saunders, 618–9.
Jovankovičová A, Jakubíková J, Durovčíková D. 2012. A case of Klippel-Feil syndrome with congenital enlarged Eustachian tube. Int J Pediatr Otorhinolaryngol 76:596–600.
Juberg RC, Gershanik JJ. 1976. Cervical vertebral fusion (Klippel-Feil) syndrome with consanguineous parents. J Med Genet 13:246–9.
Kerai S, Saxena K, Taneja B. 2014. Klippel-Feil syndrome and neuraxial anaesthesia. Indian J Anaesth 58:341–3.
Klippel M, Feil A. 1912. Un cas d’absence des vertèbres cervicales, cage thoracique remontant jusqu à la base du crâne. Nouvelle Iconographie de la Salpêtrière 25:223–50.
Lewis M. 2019. Identification of Pathological Conditions in Human Skeletal Remains. 3rd Edition. London: Academic Press, 615–37.
List CF. 1941. Neurologic syndromes accompanying developmental anomalies of occipital bone, atlas and axis. Arch Neurol Psychiatry 45:577–616.
Lowry RB, Jabs EW, Graham GE, Gerritsen J, Fleming J. 2001. Syndrome of coronal craniosynostosis, Klippel-Feil anomaly, and Sprengel shoulder with and without Pro250Arg mutation in the FGFR3 gene. Am J Med Genet 104:112–9.
MacCurdy GG. 1923. Human skeletal remains from highlands of Peru. Am J Phys Anthropol 6:217–330.
Macías-López MM. 2020. Malformaciones congénitas en columna vertebral y colesteatoma en una mujer embarazada del siglo III-IV d. C. hallada en San Fernando (Cádiz). Reconstrucción de su rostro. In: De Miguel Ibáñez MP, Romero Rameta A, Torregrosa Giménez P, Jover Maestre FJ (eds.), Cuidar, curar, morir: la enfermedad leída en los huesos. Universidad de Alicante. Instituto Universitario de Investigación en Arqueología y Patrimonio Histórico, 265–85.
Mahirogullari M, Ozkan H, Yildirim N, Cilli F, Gudemez E. 2006. Klippel–Feil syndrome and associated congenital abnormalities: evaluation of 23 cases. Acta Orthop Traumatol Turc 40:234–9.
Menger RP, Rayi A, Notarianni C. 2022 Jan. Klippel Feil Syndrome. [Updated 2021 Sep 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/NBK493157/ [Accessed 12.01.2022].
Martínez-Quintana E, Rodríguez-González F. 2015. Brief Report Klippel-Feil syndrome and levo-looped transposition of the great arteries. Cardiol Young 25:591–3.
Masset C. 1982. Estimation de l’âge an décès par les sutures crâniennes. Thése de Doctorat d’Etat. Lab-Anthropologie Biologique, Université Paris VII.
Matson DD. 1969. Neurosurgery of infancy and childhood. 2nd Edition. Springfield, Illinois: Charles C. Thomas, 119–21.
McGaughran J. 2004. Klippel–Feil Anomaly and Neural Tube Defects. Am J Med Genet 127A:327–8.
Menéndez-Bueyes LR, Soler-Fernández MC. 2017. Enfermedad ósea de Paget: aproximación a sus orígenes históricos. Reumatol Clin 13:66–72.
Mohamed JY, Faqeih E, Alsiddiky A, Alshammari MJ, Ibrahim NA, Alkuraya FS. 2013. Mutations in MEOX1, encoding mesenchyme homeobox 1, cause Klippel-Feil anomaly. Am J Hum Genet 92:157–61.
Naikmasur VG, Sattur AP, Kirty RN, Thakur AR. 2011. Type III Klippel-Feil syndrome: case report and review of associated craniofacial anomalies. Odontology 99:197–202.
Narang M, Goyal JP. 2006. Uncommon manifestations of Klippel Feil syndrome. Indian Pediatr 43:265–6.
Nouri A, Martin AR, Lange SF, Kotter MRN, Mikulis DJ, Fehlings MG. 2017. Congenital Cervical Fusion as a Risk Factor for Development of Degenerative Cervical Myelopathy. World Neurosurg 100:531–9.
Ortner D. 2003. Identification of pathological conditions in human skeletal remains. San Diego: Academic Press-Elsevier. 435–43.
Pany D, Teschler-Nicola M. 2007. Klippel-Feil Syndrome in an Early Hungarian Period Juvenile Skeleton from Austria. Int J Osteoarchaeol 17:403–15.
Papa V, Varotto E, Vaccarezza M, Ballestriero R, Tafuri D, Galassi FM. 2020. The teaching of anatomy throughout the centuries: from Herophilus to plastination. Med Hist 3:69–77.
Papagrigorakis MJ, Synodinos PN, Daliouris CP, Metaxotou C. 2003. De novo inv(2) (p12q34) associated with Klippel-Feil anomaly and hypodontia. Eur J Pediatr 162:594–7.
Papathanasiou A. 2005. Health status of the Neolithic population of Alepotrypa Cave, Greece. Am J Phys Anthropol 126(4):377–90.
Parekh SG, Donthineni-Rao R, Ricchetti E, Lackman RD. 2004. Fibrous dysplasia. J Am Acad Orthop Surg 5:305–13.
Piagkou M, Skotsimara G, Repousi E, Paraskevas G, Natsis K. 2013. Enlarged parietal foramina: a rare finding in a female Greek skull with unusual multiple Wormian bones and a rich parietal vascular network. Anat Sci Int 88:175–80.
Raus I, Coroiu RX. 2016. Mc-Albright syndrome: association of fibrous dysplasia, café-au-lait skin spots and hyperthyroidism-case report. Clujul Med 89:559–64.
Renaud A, Aucourt J, Weill J, Bigot J, Dieux A, Devisme L, Moraux A, Boutry N. 2013. Radiographic features of osteogenesis imperfect. Insights Imaging 4:417–29.
Resnick D, Kransdorf MJ. 2006. Huesos y articulaciones en imágenes radiológicas. 3rd edition. Madrid: Elsevier.
Resnik D, Niwayama G: Paget’s disease. 1998. In: Resnik D, Niwayama G, editors. Diagnosis of bone and joint disorders. Philadelphia: W.B. Saunders Company, 2127–70.
Roberts T, Stephen L, Beighton P. 2013. Cleidocranial dysplasia: a review of the dental, historical, and practical implications with an overview of the South African experience. Oral Medicine 115:46–55.
Roodman GD, Windle JJ. 2005. Paget disease of bone. J Clin Invest 115:200–8.
Russel SA: Skeletal Abnormalities. 2014. In: Coady AM, Bower S, editors. Twining’s Textbook of Fetal Abnormalities. Amsterdam: Elsevier, 417–50.
Sirico A, Maruotti GM, Martinelli P, Lanna M, Anfora R, Setaro A, Sala C. 2015. Airway management with McGrath Series 5 video laryngoscope in a woman with Klippel-Feil syndrome requiring urgent caesarean section. Int J Obstet Anaesth 24:286–8.
Strax TE, Baran E. 1975. Traumatic quadriplegia associated with Klippel-Feil syndrome: discussion and case reports. Arch Phys Med Rehabil 56:363–5.
Tachdijian MO. 1990. Pediatric orthopedics, Second Edition. Philadelphia: W.B. Saunders Company, 128.
Taylor-Martínez MA, Villanueva-Castro E, Muñoz-Romero I, De Leo-Vargas R. 2019. Síndrome de Klippel-Feil tipo 3. An Med (Mex) 64:221–4.
Thompson EM, Baraitser M, Hayward RD. 1984. Parietal foramina in Saethre-Chotzen syndrome. J Med Genet 21:369–72.
Toker S, Kilincoglu V, Unay K, Erturer E, Taser F, Gulcan E, Ilhan D. 2009. Klippel–Feil syndrome with osteopoikilosis in a young lady and her four female relatives with osteopoikilosis. Clin Rheumatol 28:235–8.
Urunuela G, Alvarez R. 1994. A report of Klippel-Feil Syndrome in Prehispanic remains from Cholula, Puebla, Mexico. J Paleopathol 6:63–7.
Varotto E, Milanese M, Tognotti E, Caramella D, Montella A, Bandiera P. 2020. Klippel-Feil Syndrome in an ancient Sardinian population (16th century AD) – A paleopathological study of four cases from the S. Michele cemetery in Alghero. In: Spani G, Varotto E, editors. Malattie e medicina tra letteratura, storia e antropologia. Holden, Massachusetts: Quod Manet, 139–53.
Vujasinovic Stupar N, Pavlov-Dolijanovic S, Hatib N, Banko B, Djukic M, Nikolic Jakoba N. 2015. Multiple Major and Minor Anomalies Associated with Klippel-Feil Syndrome: A Case Report. Arch Rheumatol 31:82–6.
Warner WC. 1998. Pediatric Cervical Spine. In: Canale ST, editor. Campbell’s Operative Orthopaedics. 9th Edition. St Louis: Mosby, 2815–47.


Received: 23.04.2022; Revised: 10.05.2022; Accepted: 18.05.2022